




Genome engineering or gene editing is a term in increasing use in our lexicon, particularly since the awareness that technologies based on prokaryotic CRISPR, an inherent part of a bacteria’s defence system against viral invasion, have therapeutic potential (Fig 1). The first in vivo CRISPR-based gene treatment for an inherited retinal disease is about to commence. So, what is it exactly, what could it be used for and, more importantly, what are its limitations?
What is CRISPR?
CRISPR refers to ‘clustered regularly interspaced short palindromic repeat’ and has a CRISPR associated protein (Cas). Initially identified in the E. coli genome, this repeat structure with adjacent genes encoding the Cas protein, appears to have developed in response to viruses integrating their genome into bacteria¹. The Cas proteins aid viral DNA to incorporate near the repeat structure and can then easily identify, target and subsequently destroy the viral DNA. Although other genome engineering tools existed before CRISPR/Cas, such as zinc-finger nucleases (ZFNs) and transcription activators like effector nucleases (TALENS), these are protein-directed DNA editing techniques. CRISPR/Cas technology’s exponential rise is due to the relative ease of application as it is a nucleotide-directed system and can be used in eukaryotic cells.

Fig 1. Nature cover March 2014 showing the excitement surrounding CRISPR
The CRISPR system can be likened to a guided missile: the CAS enzyme is the warhead, kept under control by being coupled to a guide RNA (sgRNA), which is the targeting mechanism. The sgRNA keeps the Cas protein on a leash until it finds the spot in the DNA that it is programmed to match. Once coupled, the Cas endonuclease inserts a cut or break in the double stranded DNA. The cell’s ability to repair itself is then manipulated and, via homology-directed repair (HDR), takes the two ends and joins them together, but a single nucleotide can be changed, either repairing or creating a mutation of interest. The other repair mechanism, non-homologous end joining (NHEJ) can insert or delete extra nucleotides, often resulting in gene knockouts (Fig 2).
A simple analogy would be using ‘find and replace’ on your word processor. The word of interest is highlighted by the system and replaced by the preferred word of choice. In effect, an abnormal gene sequence, ie. a disease-causing mutation, can be ‘found’ and ‘replaced’ by the normal coding sequence. Alternatively, a gene with aberrant function can be knocked out or turned off.
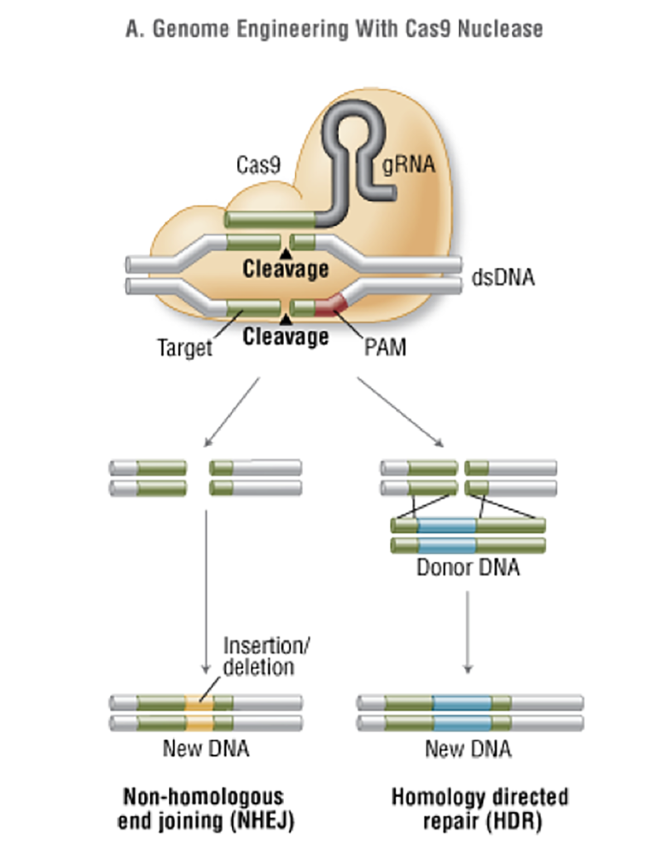
Fig 2. CRISPR/Cas Homology-directed repair and non-homologous end joining (New England Biolabs 2017)
How CRISPR is used
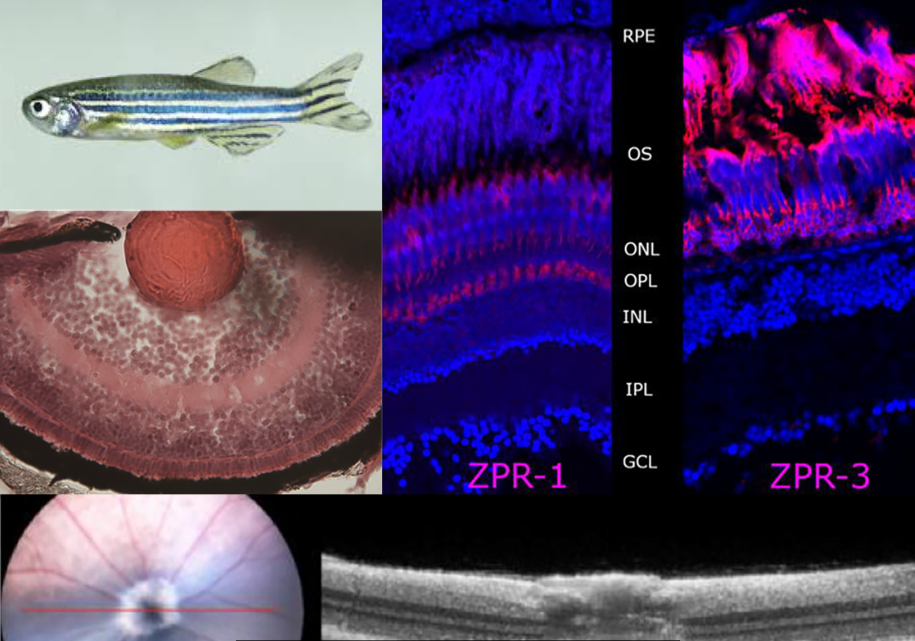
CRISPR/Cas technology has been invaluable as a tool to help understand eye disease. Using animal models such as mice or zebrafish (Fig 3), a mutation of interest can be inserted or a gene knocked out to study the functional and structural effects on the regions of interest, eg. photoreceptors. Once a model is established and disease pathogenesis more clearly understood, this animal model can be used to try therapeutic interventions, either pharmacologic or gene based, to ‘rescue’ the phenotype. Using this technology in our laboratory at the University of Auckland, medical student Micah Rapata successfully used CRISPR to create a PDE6B zebrafish mutant. There is a common founder mutation in this gene, causing autosomal rod cone degeneration in the New Zealand Māori population². Further work on this zebrafish model of retinal degeneration will help us to characterise disease progression. It can then be used to study the effect of therapeutic interventions – drugs targeting the pathways perturbed by PDE6B mutations.
Similarly, this can be applied in the lab in vitro, the so-called ‘disease in a dish’. Stem cells, either embryonic or induced pluripotent stem cells (iPSC), reprogrammed to pluripotency from a differentiated cell such as a fibroblast, can be similarly ‘edited’ to model a disease or to determine the functional implication of a new mutation. Patient-derived fibroblasts, following differentiation back to iPSCs, can then be edited to ‘repair’ a mutation.
The eye is an organ that in theory lends itself well to delivery of gene therapy and gene editing tools because of its the enclosed structure and relatively small size. The presence of the blood retinal barrier also prevents unintended systemic side effects and the ability to view and monitor both functional and structural changes from intervention. There have been models using CRISPR for age-related macular degeneration (AMD), developmental and acquired glaucoma, inherited retinal dystrophies, ciliopathies, and corneal dystrophies³.
With traditional gene therapy, a gene is added into the cell, regardless of its location, but the faulty gene is still present. In autosomal recessive or X-linked disease, this replacement may be sufficient for a therapeutic effect. In autosomal dominant diseases, where the faulty gene is still present, this approach is not effective, thus this is a potential area where gene editing, such as CRISPR/Cas, can be effective.
Mutations in the rhodopsin (RHO) gene are one of the more common causes of autosomal dominant rod cone retinal dystrophy, but a gene replacement therapy would not only require replacing the gene, but also silencing the mutant copy. However, using CRISPR/Cas in a rat model with the S334Ter Rho mutation, Bakondi et al were able to prevent further retinal degeneration⁴. Others have similarly demonstrated slowed retinal degeneration and improved visual function in transgenic rodent models targeting the human RHO P23H mutation⁵.
Bassuk et al took fibroblasts cultured from a skin punch biopsy of an XLRP patient and transduced them to produce iPSCs carrying the patient c.3070G>T mutation. The iPSCs were transduced with CRISPR gRNAs, Cas9 endonuclease and a donor homology template. They demonstrated 13% of RPGR gene copies showed mutation correction and conversion to the wild type allele⁶.
In neovascular AMD, a non-hereditary eye disease, CRISPR/Cas gene editing has been used to target the VEGF system, summarised in³. One study targeting the VEGF receptor (VEGFR2) reduced its expression by 80% in vitro, but in a murine model this fell to only 30%⁷.
This year, the first in vivo CRISPR-based therapy, in a phase 1/2A dose escalation study is due to start for early onset Leber congenital amaurosis, due to mutations in the CEP290 gene. The lab science for this, using CRISPR/Cas9, removed a de novo splice donor site caused by the most common CEP290 mutation in intron 26, c.2991+1655A>G, which results in a premature termination of the protein. Gene editing with CRISPR/Cas9, both in vitro and in a murine model, led to normal mRNA processing being restored (clinicaltrials.gov NCT03872479). The drug (AGN-151587(EDIT-101) is sponsored by Editas and Allergan and is delivered by a sub-retinal injection.
Another drug for this same condition and mutation uses an intravitreal injection of antisense oligonucleotide injection, Sepofarsen (QR110). This has completed Phase1/2 trials. Interim results reported show that in the majority of patients, a substantive overall improvement in vision was observed in the treated eye after three months, as measured by BCVA or the ability to complete mobility courses. These results were also supported by an improved visual field stimulus test and a reduction in nystagmus, with an open label extension study, Insight, for phase 1/2 participants to continue the drug. Researchers are currently recruiting for a phase 2/3 study, Illuminate.
Comparing two different modes of genetic intervention - antisense oligonucleotides vs CRISPR/Cas - for one disease will be interesting, though it could be seen as an expensive duplication of a treatment for a rare disease.
CRISPR concerns
There are areas of concern, however, with the CRISPR/Cas system. A major issue is that off- target mutations may be created, away from the intended target, and that not all mutations or gene areas of interest can be easily targeted. Similarly, the efficiency may be low, that is, the number of copies of a gene in which the gene editing occurred.
The major concerns, however, are ethical. If it is possible to edit the human genome, can it be used for enhancement? IQ, eye colour, stronger bones? Which are ethical and which are not? At the end of 2018, Chinese researcher He Jiankui revealed to a shocked world, that he had edited the genome of twin girls to confer resistance to HIV infection.
While implanting gene-edited embryos is banned in most countries, it would appear the genie is out of the bottle.
References
Dr Andrea Vincent is associate professor in the Department of Ophthalmology at the University of Auckland and a consultant ophthalmologist practicing at Greenlane Clinical Centre and at Retina Specialists. Her research focus is on the causes of inherited eye disease, particularly paediatric and retinal disease.