







Developing new drugs for the treatment of ocular diseases can be a time-consuming and costly undertaking. The trip from laboratory to market can take anywhere between 10 to 20 years and will cost around US$1 billion.
Fig 1 outlines a breakdown of the 2013 United States Food and Drug Administration (FDA) Investigational New Drug Application (IND) filing fees, estimated costs for each FDA approval phase and the likelihood of advancement between phases. As can be seen, most of the money is spent during the drug discovery and pre-clinical phases, with only five out of initially 5,000-10,000 screened compounds advancing into clinical trials, of which only one may eventually be approved. The initial discovery and development phase can take anywhere between three to six years while the majority of time is taken up by clinical trials (six to seven years), before submitting a New Drug Application (NDA) to the FDA (Fig 2). Overall, the drug development process can be divided into five steps1.
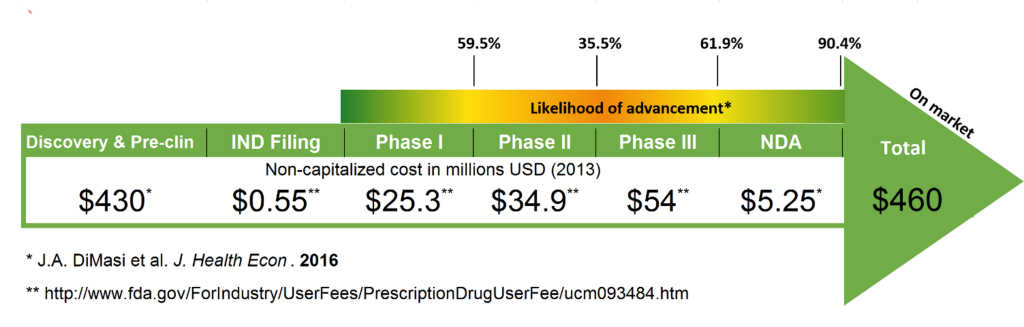
Fig 1. Estimated non-capitalised costs for new drug development and advancement between FDA approval phases2
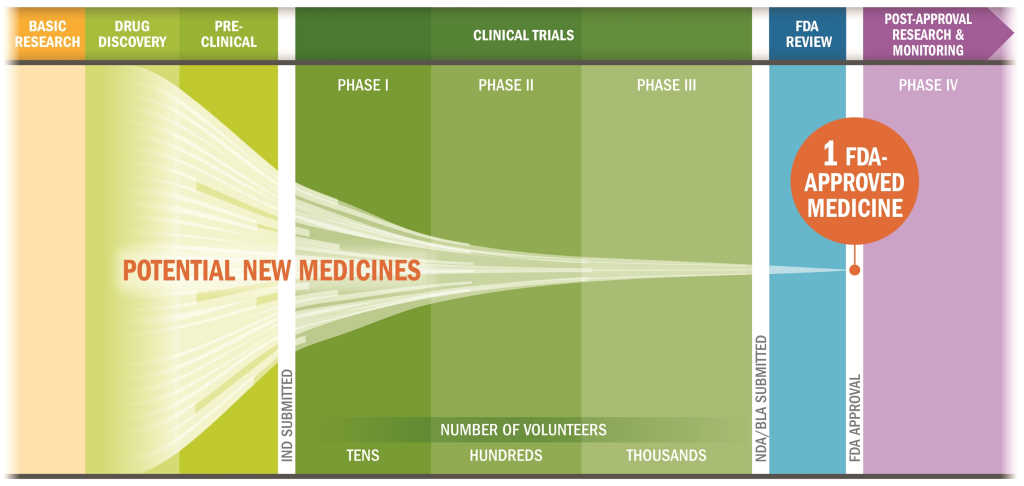
Fig 2. Drug research and development process (IND: Investigational New Drug Application; NDA: New Drug Application; BLA: Biologics License Application)3
Step 1 - Discovery and development
Research for a new drug begins in the laboratory
Novel drugs are often discovered after new insights into a disease process have been gained allowing the researcher to design or screen for suitable molecules. Discoveries can also be made when an existing treatment shows unexpected effects. For example, patients on bimatoprost (Lumigan) eye drops, a prostaglandin analogue used in the treatment of open-angle glaucoma and approved by the FDA in 2001, experienced growth of longer, thicker and darker eye lashes resulting in the approval of Latisse for cosmetic purposes in 2008. Repurposing of an already existing drug with proven safety is also generally much faster and less costly.
While thousands of compounds may be potential candidates for drug development, only a small number actually proceed to preclinical studies. Thus, once a promising candidate has been identified, further experiments are performed to elucidate the drug pharmacokinetics, the exact mechanism of action, the best dose and route for administration as well as any potential adverse effects. Only if there is no toxicity and the candidate shows clear advantages over current drugs on the market, ie. a different mechanism of action, less adverse effects or less frequent dosing, should it proceed into preclinical studies.
Step 2 - Preclinical research
Drug undergoes laboratory and animal testing
Before advancing to clinical trials, researchers must confirm there is no potential for the new molecule to cause serious harm using in vitro (cell culture) and in vivo (animal) studies. Such studies must be performed under good laboratory practice (GLP) guidelines, setting minimum requirements for personnel, equipment, protocols, operating procedures, reports and quality assurance. Pre-clinical studies are generally small with often only six replicates per treatment group. However, they must still provide sufficient detail on dosing and toxicity before the candidate can move into clinical trials.
One of the greatest challenges for translation of a drug from the laboratory bench into humans remains the poor correlation of pre-clinical data with clinical trial results often due to the different anatomy and physiology of most animal eyes. Moreover, the majority of animal disease models are acute and very homogenous while the human condition is often chronic and can vary significantly between patients. It is therefore no surprise that a number of molecules having shown great efficacy in animal studies end up failing in clinical trials.
Step 3 - Clinical research
Drug’s tested in humans to prove safety and efficacy
While pre-clinical research can answer basic questions about a drug’s safety and efficacy, it is not a substitute for studying the drug’s interactions with the human body. However, before being allowed to start clinical trials, an IND has to be submitted, including animal study and toxicity data, manufacturing information, clinical trial design, data from any prior human research and information about the investigator. The FDA then has 30 days to review and respond to the IND submission. Clinical trials are designed to answer specific research questions related to the new drug and a number of critical aspects such as patient selection criteria, participant numbers, study duration, data to be collected and how these will be analysed, need to be carefully considered. Typically, clinical trials follow a series from early, small-scale, Phase 1 studies to late-stage, large scale, Phase 3 studies followed by even larger Phase 4 trials after market approval (Table 1).
Table 1 Clinical trial phase overview1
|
Phase |
Number of volunteers |
Duration |
Purpose |
% of drugs moving into next phase |
|
1 |
20-100 (often healthy) |
Several months |
Safety and dosage |
70% |
|
2 |
100-300 (diseased) |
Several months to 2 years |
Efficacy and side effects |
33% |
|
3 |
300-3,000 (diseased) |
1- 4 years |
Efficacy and side effects |
25-30% |
|
4 |
Thousands (diseased) |
Several years |
Long-term safety and efficacy |
NA |
Step 4 - FDA review
Thorough examination of all submitted data
If pre-clinical and clinical research has shown a drug is safe and effective for its intended use, an NDA is submitted to the FDA, whose review team thoroughly examines all the submitted data before making a decision. The NDA has to contain everything about the drug, from preclinical data to Phase 3 trial results, as well as information required for marketing such as the proposed labelling and directions of use. If the application is deemed complete during the initial screening, the FDA has six to 10 months to make a decision on whether to approve the drug. In most cases, however, remaining issues such as addressing specific questions based on existing data or providing additional data, need to be addressed before a drug can be approved. A recent example in the ophthalmic field includes the rejection of Dextenza (Ocular Therapeutix), an intracanalicular dexamethasone insert intended for the treatment of ocular pain and inflammation following ophthalmic surgery, due to deficiencies in manufacturing processes and analytical testing. However, the company aims to re-file the NDA in 2018 after addressing the raised concerns.
Step 5 - Post-market safety monitoring
FDA monitoring of drug safety once available for the public
Although clinical trials provide important information on a drug’s efficacy and safety, it is impossible to have complete information about the drug’s safety at the time of approval. Despite the rigorous steps taken during the drug development process, limitations exist. Therefore, the true picture of a product’s safety generally evolves over months and even years after marketing. The FDA reviews any problem reports and can decide to add cautions to the usage information or even withdraw the product in case of more serious issues.
Generic drugs, new drug delivery systems and natural health products
Newly approved drugs are generally patent protected which means only the sponsor has the right to market the drug. However, once the patent expires - typically 20 years from the earliest application filing date - other drug manufacturers can develop a generic version, which must have the same dosage form, strength, safety, quality, performance and intended use. If all of these are adhered to, generic drug manufacturers only have to perform bioequivalence studies instead of conducting lengthy and costly clinical trials.
Already approved drugs may also be combined with a new delivery system such as a sustained release implant, as is the case for the Ozurdex dexamethasone implant. However, although safety and efficacy of the incorporated drug have already been demonstrated in numerous clinical trials, the combination product is considered a new entity and an additional IND has to be filed before conducting further clinical trials. These may be less extensive, but still mean many years of development and additional dollars, thus only combination products that show clear advantages over already approved formulations should be further investigated.
In comparison to many other countries, natural health products are currently unregulated in New Zealand. While the Government and the Green Party announced plans to develop a regulatory scheme for natural health products to reduce risks associated with unsafe ingredients, uncontrolled health claims and poor manufacturing in 2009, the Natural Health Products Bill has not been reinstated by the new government so far.
Final remarks
Looking at FDA approval numbers between 2012 and 2017 (584 in total), ophthalmic products only made up a very small percentage (<5%) with a decline from 31 (2006-2001) to 25 (2012-2017) seen. However, with three NDA submissions already in the pipeline for 2018 and a number of important clinical trial read-outs expected, we may see a surge of ophthalmic product approvals in the near future.
References
About the Author
Dr Ilva Rupenthal is a senior lecturer in the University of Auckland’s Department of Ophthalmology and the director of the Buchanan Ocular Therapeutics Unit (www.botu.nz), which aims to translate ocular therapeutic related scientific research into the clinical setting.